Machine learning--assisted CO2 utilization in the catalytic dry reforming of hydrocarbons: Reaction pathways and multicriteria optimization analyses
 Graphical Abstract
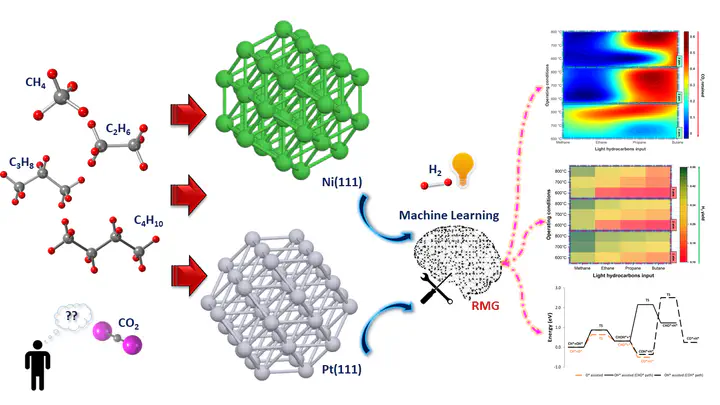
Graphical AbstractAbstract
The catalytic dry reforming (DR) process is a clean approach to transform CO2 into H2 and CO-rich synthetic gas that can be used for various energy applications such as Fischer–Tropsch fuels production. A novel framework is proposed to determine the optimum reaction configurations and reaction pathways for DR of C1-C4 hydrocarbons via a reaction mechanism generator (RMG). With the aid of machine learning, the variation of thermodynamic and microkinetic parameters based on different reaction temperatures, pressures, CH4/CO2 ratios and catalytic surface, Pt(111), and Ni(111), were successfully elucidated. As a result, a promising multicriteria decision-making process, TOPSIS, was employed to identify the optimum reaction configuration with the trade-off between H2 yield and CO2 reduction. Notably, the optimum conditions for the DR of C1 and C2 hydrocarbons were 800°C at 3 atm on Pt(111); whereas C3 and C4 hydrocarbons found favor at 800°C and 2 atm on Ni(111) to attain the highest H2 yield and CO2 conversion. Based on the RMG-Cat (first-principle microkinetic database), the energy profile of the most selective reaction pathway network for the DR of CH4 on Pt(111) at 3 atm and 800°C was deducted. The activation energy (Ea) for CH bond dissociation via dehydrogenation on the Pt(111) was found to be 0.60 eV, lower than that reported previously for Ni(111), Cu(111), and Co(111) surfaces. The most endothermic reaction of the CH4 reforming process was found to be C3H3* + H2O* ↔ OH* + C3H4 (218.74 kJ/mol).
Juin Yau Lim
Ph.D, M.Eng, AMIChemE (he/him/his)
Passionate sustainable practitioner that seeks solutions with modern approaches.